The value of a Quality Target Product Profile
Clinical trials make up a substantial portion of the overall drug development costs and pharmaceutical companies are intensely focused on ensuring that the trial is well-designed and developed. The development of the actual drug product, the Chemistry, Manufacturing, and Controls (CMC) section of Investigational Medicinal Products Dossiers (IMPDs) or Investigational New Drug (IND) applications, is often not given the same level of attention.
By the LINK Medical Regulatory team

Lone Dyrby, Regulatory Director
The lack of attention when it comes to sufficient planning, developing, and verifying of the actual CMC section of any pharmaceutical project is most common during the very early development phase; from pre-clinical to clinical phase 2A.
To gain a competitive clinical edge, pharmaceutical companies may look to do the most minimal CMC work required while still maintaining an acceptable level of control, as required by the competent authority. However, the question that arises is whether such decisions truly accelerate the overall development project.
Drugs and biopharmaceuticals approved by the FDA, where Target Product Profiles (TPPs) are mentioned, are associated with more efficient regulatory review times, perhaps because of increased planning or the TPP promotes well-organised regulatory dialogue. A well-defined Quality Target Product Profile (QTPP) that considers key CMC issues is one potential way to obtain early regulatory approval.
CMC activities during the development
CMC activities include establishing manufacturing processes and product characteristics, defining product testing methods to ensure the drug product’s safety and efficacy, and consistency between batches.
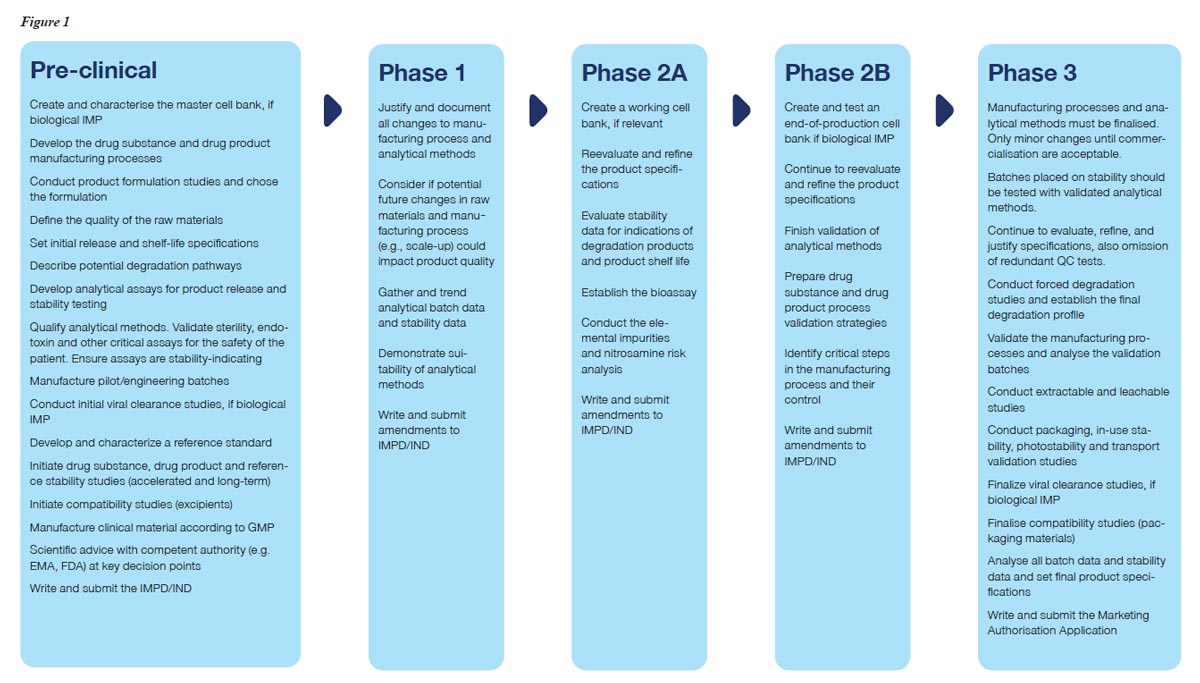
The regulators expect there is consistency between the product tested during the clinical trials and commercialised batches produced years later. To minimise the risk of later-stage failure, several key CMC activities must be considered during the development of biopharmaceuticals, see Figure 1.
Read more and register to our next webinar: The Importance of Regulatory CMC Strategy During Early Development

The main part of the CMC activities defining the quality characteristics of the commercial product is performed during early development, that is in the pre-clinical phase and further developed until Phase 2AB. It is during these phases of development that the quality characteristics of a drug product ideally will be achieved to ensure the desired quality, considering the safety and efficacy of the drug product.
Establishing the design criteria for the different CMC activities in a structured and strategic way minimises the risk of repeating studies or delaying product development.
The value of a Quality Target Product Profile (QTTP) in early development
The QTPP is an overview of all the elements during the product development process that have an impact on the quality, safety, and efficacy of a product in a given clinical indication. The QTPP does not only focus on CMC issues but should also cover the specific product design, mode of action of the product, intended clinical use, dosing, route of administration, and so on.
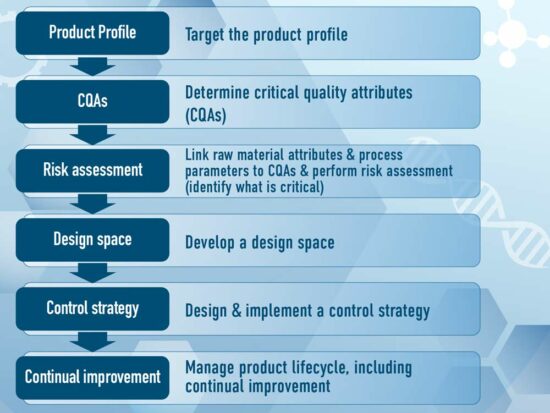
The QTPP is part of the ICH Q8 (R2) enhanced approach as a more systematic approach to development including, for example, incorporation of prior knowledge, results of experimental studies using design of experiments or multivariate data analysis, the establishment of a control strategy, use of quality risk management, and use of knowledge management throughout the lifecycle of the product, see Figure 2.
Figure 2 ICH Q8(R2) Enhanced Approach
The definition of a QTPP early in the development of a new biopharmaceutical can be useful in identifying potential areas of CMC risks and designing adequate (and inexpensive) remediation strategies.
For example, product instabilities in the form of aggregates, impurities, or degradation products are often responsible for the incidence of immunogenic responses in patients. These criteria, therefore, are often closely linked to specific quality attributes, namely, aggregation levels (as well as other product impurities), product yields, and biological activity. From these quality attributes, several design criteria can be used to define a suitable developability program.
The design criteria could include increasing product stability, reducing aggregation, maintaining adequate productivity, and achieving all these requirements whilst maintaining acceptable affinity to the target.
The design plan should include a risk assessment, mapping areas of the molecule potentially responsible for the observed behaviour, and the introduction of a mitigation plan that could involve the substitution of key residues in the molecule to improve the required parameters.
Finally, the resulting product candidates should be assessed using relevant experimental techniques to determine whether the remediation plans satisfy the requirements for the product. In this example, the QTPP defines the target as “minimise resistance to the drug” that identifies the quality attribute “low immunogenicity” and points out that the development of the drug should focus on minimising aggregation.
Building up the QTPP should start at the research phase and continue up to the Marketing Authorisation Application (MAA). The QTPP is a living document. Throughout the development process, when changes are done and new data are accumulating, the information should be used to update the QTPP to assess the need for comparability studies and to evaluate possible gaps that may need to be filled. If it is put together properly and regularly updated, it provides the skeleton for the entire CMC module of the IMPD and the end target – the MAA.
How can LINK Medical support establishing a QTPP?
LINK Medical’s Regulatory team of CMC experts can assist with identifying gaps in the regulatory CMC documentation and together with the client at a QTPP workshop identify the “minimal acceptable results” and “ideal results” for each CMC activity.
A QTPP workshop held early in development can identify regulatory CMC gaps that could have resulted in delayed initiation of Phase 1 clinical trial had the gaps not been identified.
Register to our Webinar
The Importance of Regulatory CMC Strategy During Early Development
Accelerate development using a Quality Target Product Profile
Having the right Chemistry, Manufacturing, and Controls (CMC) documentation throughout drug development is essential for avoiding pitfalls, ensuring an efficient regulatory review process, and achieving the goals for your product. The key is an early development regulatory strategy that includes CMC.
Join our regulatory CMC webinar to strengthen your knowledge of how regulatory CMC strategies can accelerate the drug development process and become familiar with strategic tools such as the Quality Target Product Profile (QTPP).